

Translate this page into:
Mitochondrial Disease Masquerading as Recurrent Encephalopathy

Abstract
A young male who was a diabetic presented with gradually progressive hearing loss, recurrent episodic behavioral abnormality, easy fatigability, proximal weakness, and seizures. The patient was evaluated in detail to find out the etiology of encephalitis. The serum autoimmune encephalitis panel was negative. On magnetic resonance imaging of the Brain, there was temporoparietal T2 and fluid-attenuated inversion recovery hyperintensity. In the magnetic resonance spectroscopy study, a lactate peak was found. So, the patient was evaluated for mitochondrial illness mitochondrial encephalopathy lactic acidosis with stroke-like symptoms) and it was confirmed by a genetic study. This case illustrates that though it is a rare disease, always a strong clinical index of suspicion is required for the diagnosis of mitochondrial disorders in case of multisystem involvement so that diagnosis would not be missed.
Keywords
Genetic study
Lactate peak
MELAS

Introduction
Mitochondrial disorders are a rare group of genetic diseases with maternal inheritance.[1] These are caused by a wide spectrum of mutations in genes encoded by the nuclear or mitochondrial genomes.[1] It affects multiple systems of the body. The disease activity, penetrance, and severity are related to clinical presentation and characteristic imaging findings.[2] There is no specific treatment for these disorders. We are reporting a case of mitochondrial encephalopathy lactic acidosis and stroke-like symptoms (MELAS) that is not very common and could have been missed if not suspected.
Case Report
A 21-year-old male presented with exercise intolerance and gradually progressive hearing loss for 2 years, recurrent episodic behavioral abnormality for 2 months, and 1 episode of seizure. There was no associated fever, headache, tinnitus, nausea, or vomiting. He was a diabetic detected 2 years back and was on insulin. He also developed forgetfulness for the last 1 month affecting mainly recent memory. He was treated as a case of viral encephalitis after the first episode of seizure with antivirals and antiepileptics. But the behavioral abnormalities persisted, so he was evaluated again for autoimmune encephalitis and vasculitis. He had a history of exercise intolerance and fatigability while doing workouts at the gym. There was no h/o dysphagia, ataxia, dizziness, double vision, or bowel or bladder abnormalities. He was educated up to the 12th standard. The perinatal period was uneventful. No h/o developmental delay. No h/o similar illness in siblings or any other family members. No h/o diabetes in the mother. On examination, there was profound hearing loss and mild weakness of proximal muscles.
All routine investigations were normal except hemoglobin A1c which was 7.9.csf autoimmune encephalitis panel and viral meningitis and encephalitis panel was negative. Antinuclear antibody profile, c antineutrophil cytoplasmic antibodies (cANCA), and pANCA were all negative. L-lactate was high 28.92 (4.5–19.2) mg/dL. Serum CPK 36U/L. Serum CKMB-45 U/L. Serum lactate dehydrogenase 332U/L. Audiometry revealed bilateral severe sensorineural hearing loss.
Magnetic resonance imaging (MRI) brain showed cortical and subcortical fluid-attenuated inversion recovery and T2 hyperintensities at right temporal and left temporoparietal region [Figures 1 and 2]. Electroencephalogram showed epileptiform activity from the left temporal region with generalized background slowing. Magnetic resonance spectroscopy (MRS) shows presence of lipid lactate peak within the lesion [Figure 3]. So, with strong clinical suspicion genetic study was advised for mitochondrial mutation detection comprehensive panel that showed mutation in A12397G. A diagnosis of MELAS was made. The patient was discharged with tab oxcarbazepine, L-carnitine, Coq10, insulin to control blood sugar.
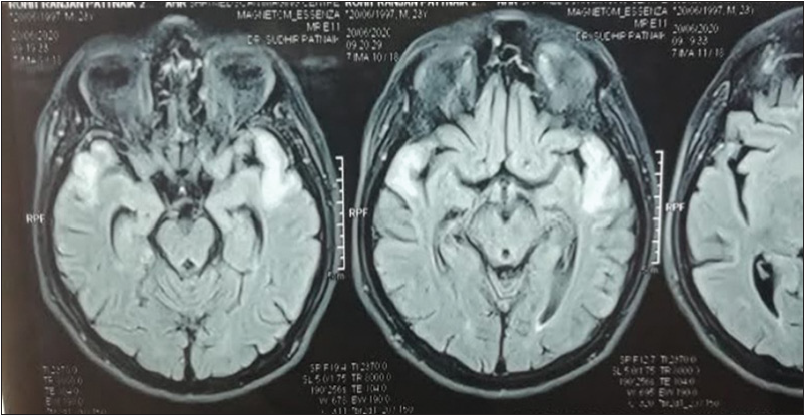
- Fluid-attenuated inversion recovery hyperintensity in right temporal and left temporal region.
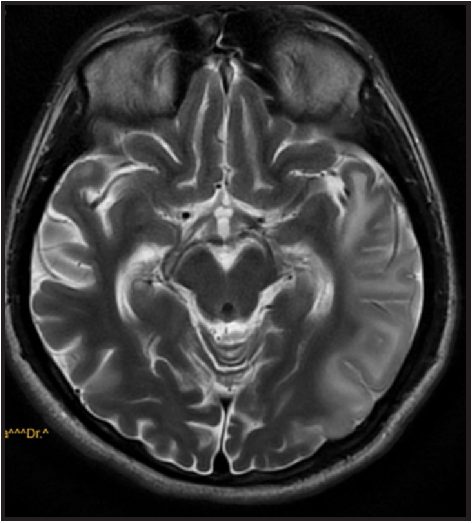
- T2 hyperintensity over right temporal and left temporoparietal region.
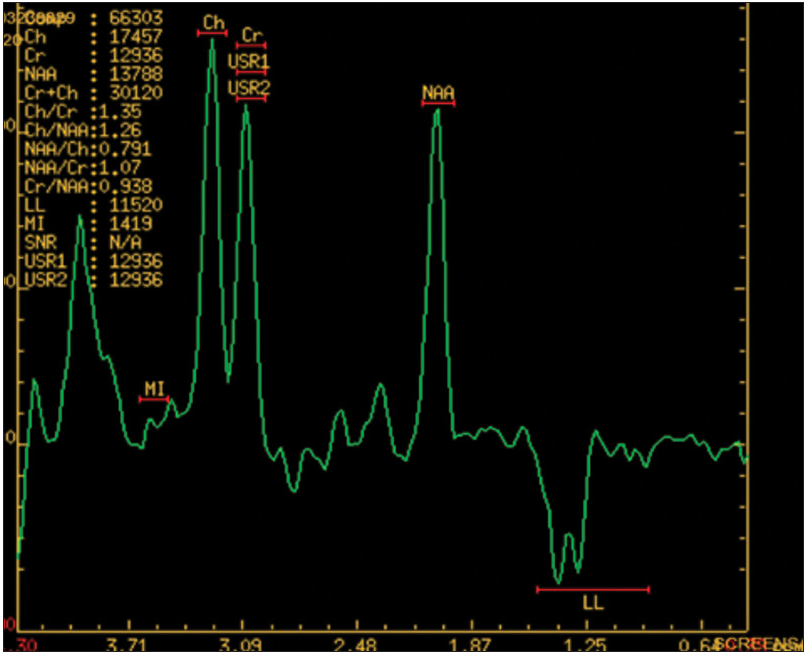
- MRS-LACTATE peak.
Discussion
The patient mainly presented with recurrent encephalopathy and seizures. So, the differential diagnosis was viral encephalitis, autoimmune encephalitis, primary or secondary central nervous system vasculitis and lymphoma. We excluded all one by one. Presence of diabetes, sensorineural hearing loss, waxing waning course, typical MRI and MRS findings patient was evaluated for mitochondrial disease. The percentage level of mitochondrial DNA pathogenic variants may vary among individuals of the same family and even among organs and tissues of same individual.[3,4] Stoke-like episodes in MELAS can have focal neurologic deficits due to localized lesion in brain; episodes are often associated with seizures; the lesions do not follow any vascular territory with predilection for posterior brain region and gradually spread to involve wider areas of brain. The neurological findings may disappear along with radiological findings only to recur later.[5]
Conclusion
Strong clinical suspicion is required for diagnosis. Mitochondrial dysfunction should be considered in the differential diagnosis of any progressive multisystem disorder. With more than 1,000 nuclear genes encoding mitochondrial proteins, the molecular diagnosis can be challenging.
Funding
None.
Conflict of interest
None declared.
References
- Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133-58.
- [CrossRef] [PubMed] [Google Scholar]
- Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes-MELAS syndrome. Ochsner J. 2017;17:296-301.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MELAS: mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. Brain Nerve. 2017;69:111-7.
- [CrossRef] [PubMed] [Google Scholar]




