

Translate this page into:
Pituitary Hyperplasia in a Boy with Van Wyk–Grumbach Syndrome due to Long-standing Hypothyroidism
Corresponding Author: Shakil Shaikh, Resident Medical Officer, Department of Pediatric Medicine, Bai Jerbai Wadia Hospital for Children, Mumbai, Maharashtra, India, Phone: +919757481184, e-mail: drshakilsshaikh@rediffmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
How to cite this article: Shaikh S, Joshi R. Pituitary Hyperplasia in a Boy with Van Wyk-Grumbach Syndrome due to Long-standing Hypothyroidism. Int J Recent Surg Med Sci 2016;2(1):44-46.
Abstract
Van Wyk–Grumbach syndrome (VWGS) is characterized by juvenile hypothyroidism, delayed bone age, and pseudo-precocious puberty. Primary hypothyroidism in the juvenile population generally leads to retardation of linear growth and delayed puberty. However, in rare conditions, children with long-standing hypothyroidism present with signs of VWGS. It can occur in both sexes. In girls, the usual presenting features are early-onset menarche and enlarged bilateral multicystic ovaries, whereas in boys it is rarely associated with testicular enlargement. We present an unusual case of an 8-year-old boy who was referred to us with a pituitary tumor (macroadenoma) and later on diagnosed as having long-standing untreated congenital hypothyroidism with testicular enlargement and pituitary hyperplasia. Eight months after thyroxine replacement therapy, repeat magnetic resonance imaging shows complete resolution of pituitary hyperplasia.
Keywords
Congenital hypothyroidism
Kocher–Debre–Semelaigne syndrome
Pituitary hyperplasia
Precocious puberty
Van Wyk–Grumbach syndrome.

INTRODUCTION
Long-standing primary hypothyroidism in children usually leads to both pubertal and growth delay, but in rare cases hypothyroidism leads to growth delay with paradoxically precocious puberty and delayed bone age. Van Wyk and Grumbach1 first described the combination of juvenile hypothyroidism, delayed bone age, and precocious puberty in 1960. Most of the Van Wyk–Grumbach syndrome (VWGS) cases were reported in prepubertal girls with early-onset menarche and enlarged multicystic ovaries, while it is very rare in boys and manifests with testicular enlargement.2 Pituitary enlargement secondary to long-standing hypothyroidism is a known but uncommon occurrence. Thyrotroph hyperplasia can result in the expansion of sella turcica and the enlargement of pituitary gland.3 We present an unusual case of an 8-year-old boy who presented with severe growth failure, global developmental delay, precocious puberty (testicular enlargement), and pituitary hyperplasia (initially diagnosed as pituitary tumor) due to long-standing untreated congenital hypothyroidism.
CASE REPORT
An 8-year-old boy presented with complaints of not gaining in height and weight since 1 year of age with global developmental delay. Antenatal and birth history was uneventful with no neonatal complication. There was no past history of irradiation, trauma, and surgery. On enquiry, history of constipation, lethargy, snoring during sleep was present. He had no family history suggestive of thyroid disease, autoimmunity, or precocious puberty. On examination pulse rate was 68/minute with respiratory rate 24/minute. He had pallor, coarse facial features with myxedematous appearance, periorbital puffiness, thick edematous lips, macroglossia, coarse brittle hair, dry rough skin, and pseudohypertrophy of calf muscles (Fig. 1). Thyroid gland was not palpable with normal oral cavity examination. Genital examination showed bilateral testicular enlargement measuring 5 mL in size (pubertal) by Prader orchidometer. Stretch penile length was 6 × 1.5 cm with absent axillary and pubic hair. Central nervous system examination revealed delayed relaxation of deep tendon reflexes. Rest of the systemic examination was normal. Signs of rickets in the form of rib beading, wrist widening, double malleoli with significant bowing of legs were present. His weight was 10 kg and height was 79 cm (both are below 3rd percentile on Indian Academy of Pediatrics growth charts). His height age and weight age was 18 months. His bone age was 3 months according to Greulich and Pyle atlas method. Hormonal investigations revealed free T3: 1.62 pg/mL [normal range (NR): 2.5–3.9] and free T4: 0.08 ng/dL (NR: 0.61–1.12), with thyroid stimulating hormone (TSH) more than 100 µIU/mL (NR: 0.35–5.5); follicle stimulating hormone (FSH): 0.07 mIU/mL (NR: 0.15–3.1); luteinizing hormone (LH): < 0.09 mIU/mL (NR: 0.7–1.3); and serum testosterone: 12.98 ng/dL. Ultrasonography neck showed small-sized thyroid gland with normal echogenicity. The referring hospital had done magnetic resonance imaging (MRI) of brain with gadolinium, which revealed well-defined T2 isointense and T1 isointense lesion measuring approximately 16.3 × 10.3 × 7.9 mm in sella with indentation of optic chiasma superiorly and was reported as pituitary macroadenoma. Based on the MRI study, the patient was suggested neurosurgical intervention. However, in view of clinical profile and hormonal investigation, we made a diagnosis of congenital hypothyroidism, and pituitary changes were attributed to compensatory hyperplasia secondary to long-standing hypothyroidism. Repeat MRI 8 months after thyroxine replacement revealed complete resolution (6.9 × 6.8 × 9.3 mm) of the enlarged pituitary mass (Figs 2A and B).
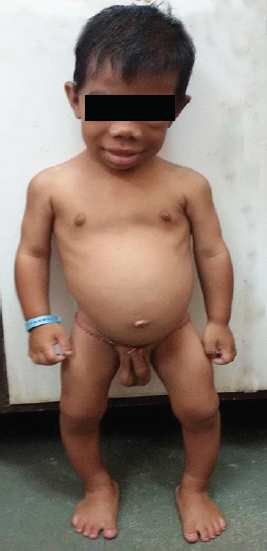
- Clinical photograph of a patient with features of hypothyroidism along with testicular enlargement

- (A) Magnetic resonance imaging of brain (sagittal section) showing pituitary hyperplasia before thyroxine treatment, and (B) complete resolution of pituitary hyperplasia after treatment (by arrow)
DISCUSSION
Finding of delayed bone age in children with precocious puberty narrows the differential diagnosis to hypothyroidism as other causes of precocious puberty present with an advanced bone age.4 This can be explained based on the fact that thyroid hormone-mediated bone maturation involves direct and indirect actions. The indirect action is mediated by the regulation of growth hormone gene expression and the insulin-like growth factor system, while T3 directly regulates the endochondral ossification and also controls chondrocyte differentiation in the growth plate both in vitro and in vivo.5 The pathophysiology of paradoxical precocious puberty with primary hypothyroidism in VWGS involves a complex interaction between different hypothalamic–pituitary hormonal axes. In the original description, Van Wyk and Grumbach1 hypothesized that there was hormonal overlap in the pituitary feedback mechanism. Hormonal overlap at the level of G-protein coupled receptors (GPCRs) due to common alpha-subunit between TSH and FSH is the underlying principle. Each acts through the transmembrane GPCRs to activate adenylate cyclase and stimulate cyclic adenosine monophosphate production.6 In addition, using recombinant tools, it has been shown that human TSH can interact with the human FSH receptor to stimulate the adenyl cyclase activity.7 In vitro experiments have demonstrated that TSH can weakly stimulate the FSH receptor without simultaneous stimulation of LH receptors explaining the low prepubertal level of LH. Therefore, high levels of TSH may act through the FSH receptor to cause gonadal stimulation and precocious sexual changes.7 In boys, severe hypothyroidism leads to over proliferation of Sertoli cells in prepubertal testis causing testicular enlargement without significant virilization. Testicular histology shows a predominance of tubular elements without elevated Leydig cell number, consistent with an FSH-mediated response.7,8 Our patient also had pseudohypertrophy of calf muscles which is described in the literature as Kocher–Debre–Semelaigne syndrome seen in long-standing hypothyroidism.9
Pituitary enlargement with long-standing profound hypothyroidism results from loss of negative feedback mechanisms and causes secondary hypertrophy and hyperplasia of the thyrotrophic cells in the anterior lobe of the pituitary gland.3 Despite progress in medical imaging, to date, even MRI cannot distinguish pituitary macro-adenoma from hyperplasia.10 Therefore, the diagnosis of thyrotrophic pituitary hyperplasia must rely heavily on the patient's clinical profile and on detailed endocrine work-up. Without the knowledge of our patient's clinical condition (hypothyroidism), the pituitary mass was initially reported as a macroadenoma. After documenting primary hypothyroidism, we considered the diagnosis of reactive pituitary hyperplasia, which was definitively confirmed by the regression of the pituitary mass on the follow-up MRI after thyroxine replacement therapy, allowing us to avoid unnecessary neurosurgical intervention.
This case highlights the need to familiarize clinicians with delayed bone age as a discriminating feature between VWGS from the other causes of precocious puberty and also about uncommon presentation of pituitary hyperplasia in untreated hypothyroidism.
Written informed consent was obtained from the parents of the patient for publication of this case report and accompanying images.
Source of support:
Nil
ACKNOWLEDGMENT
Authors are grateful to Dr. YK Amdekar, Medical Director, Bai Jerbai Wadia Hospital for Children, Mumbai for allowing us to publish this clinical brief.
Dr. Shakil Shaikh and Dr. Rajesh Joshi were involved in conceptualization of the manuscript, collecting patient data, conducting literature search, and drafting the manuscript. This was critically reviewed and approved by Dr. Rajesh Joshi. Both authors were involved in the clinical management of the patient.
Dr. Rajesh Joshi will act as the guarantor of the manuscript.
Conflict of interest:
None.
REFERENCES
- Syndrome of precocious menstruation and galactorrhea in juvenile hypothyroidism: an example of hormonal overlap in pituitary feedback. J Pediatr. 1960;57(3):416-435.
- [CrossRef] [Google Scholar]
- Current concepts in normal and abnormal puberty. Curr Probl Pediatr Adolesc Health Care. 2007;37(2):50-72.
- [CrossRef] [PubMed] [Google Scholar]
- Pituitary enlargement on magnetic resonance imaging in congenital hypothyroidism. Arch Pediatr Adolesc Med. 1996;150(6):623-628.
- [CrossRef] [PubMed] [Google Scholar]
- Primary hypothyroidism presenting as severe vaginal bleeding in a prepubertal girl. J Pediatr Adolesc Gynecol. 1997;10(1):35-38.
- [CrossRef] [PubMed] [Google Scholar]
- Interactions between GH, IGF-I, glucocorticoids, and thyroid hormones during skeletal growth. Pediatr Res. 2002;52(2):137-147.
- [CrossRef] [PubMed] [Google Scholar]
- G-protein-coupled receptors at a glance. J Cell Sci. 2003;116:4867-4869. Pt 24
- [CrossRef] [PubMed] [Google Scholar]
- A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J Clin Endocrinol Metab. 1995;80(1):276-279.
- [CrossRef] [PubMed] [Google Scholar]
- Hypothyroidism-induced macroorchidism: use of a gonadotropin-releasing hormone agonist to understand its mechanism and augment adult stature. J Clin Endocrinol Metab. 1995;80(1):11-16.
- [CrossRef] [PubMed] [Google Scholar]
- Kocher-Debre-Semelaigne syndrome: hypothyroidism with muscle pseudohypertrophy. Indian J Pediatr. 2003;70(8):671-673.
- [CrossRef] [PubMed] [Google Scholar]
- Spontaneous ovarian hyperstimulation syndrome and pituitary hyperplasia mimicking macroadenoma associated with primary hypothyroidism. World J Radiol. 2013;5(1):20-24.
- [CrossRef] [PubMed] [Google Scholar]




